%config InlineBackend.figure_formats = ['svg']
%matplotlib inline
Alanine Dipeptide in Implicit Water#
This example reproduces the results of the paper Deep Coarse-Grained Potentials via Relative Entropy Minimization [1].
This paper introduces the bottom-up coarse-graining approach of Relative Entropy Minimization (REM) to Neural Network (NN) potential models. In the example of alanine-dipeptide in implicit water, the paper compares REM to the conventional Force Matching (FM) scheme.
We outline the theoretical backgrounds of the REM and FM schemes in the toy examples to Relative Entropy Minimization and Force Matching. We refer to the original paper for a more detailed analysis of the connection between those approaches.
Problem#
Alanine dipeptide is a good test case for developing coarse-grained protein models. Torsional potentials derived from alanine dipeptide generalize well to larger amino acids [2]. Thus, this example compares two algorithms for learning Graph Neural Network (GNN) potentials based on representing the torsional preferences of alanine dipeptide.
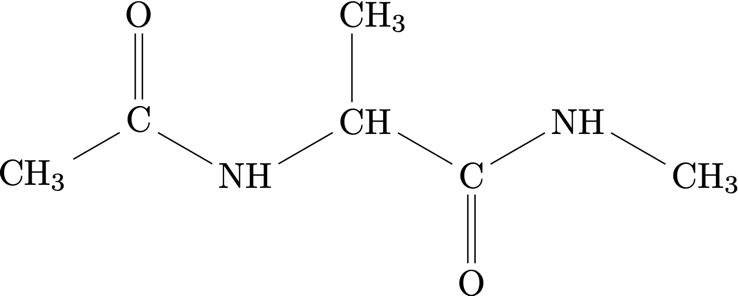
Alanine Dipeptide#
The coarse-grained model of alanine dipeptide only preserves the heavy atoms $\mathrm C$, $\mathrm N$, and $\mathrm O$ but distinguishes between carbon with different environments. Therefore, the coarse-grained model contains the five species $\mathrm{CH_3}, \mathrm{CH}, \mathrm{C}, \mathrm{O}, \mathrm{N}$. Additionally, the GNN does not explicitly consider water and should thus implicitly include the interactions with the solvent.
# Set random key and thermodynamic statepoint (300 K)
key = random.PRNGKey(21)
kT = 300. * quantity.kb
state_kwargs = {'kT': kT}
Before defining the potential model, we set up the space in which it acts. Therefore, we load an initial conformation, define a corresponding periodic space, and construct a neighbor list with a cutoff radius of $0.5~\text{nm}$.
# To construct a NN model, we first need to create a neighborlist.
# The neighborlist is necessary to construct a graph containing the environment
# of each atom.
box, r_init, _, _ = io.load_box("../_data/alanine_heavy_2_7nm.gro")
n_species = 5
r_cut = 0.5
fractional = True
displacement_fn, shift_fn = space.periodic_general(
box, fractional_coordinates=fractional)
if fractional:
box_tensor, scale_fn = custom_space.init_fractional_coordinates(box)
r_init = scale_fn(r_init)
else:
box_tensor = box
neighbor_fn = partition.neighbor_list(
displacement_fn, box_tensor, r_cut, disable_cell_list=True,
fractional_coordinates=fractional, capacity_multiplier=1.5
)
nbrs_init = neighbor_fn.allocate(r_init, extra_capacity=1)
To improve the stability of the learned potential, we employ a $\Delta$–learning approach. As a prior, we use commonly used intra-molecular potentials and repulsive non-bonded terms
where $\mathcal B, \mathcal A, \mathcal D$ are the indices of the atoms that form bonds, angles, and dihedral angles, respectively. Note also that atoms do not interact via the nonbonded term if involved in the same bond, angle, or dihedral angle. For the bonds and angles, we choose a harmonic potential $$ U_b(x) = \frac{k}{2}(x - x_0)^2, $$ where we derived the parameters $k$ and $x_0$ for each combination of species from the mean and variance of $x$ $$ x_0 = \langle x \rangle_\text{AT}, \quad k = \frac{1}{2\beta\langle(x - x_0)^2\rangle_\text{AT}}. $$ For the dihedral angles, we choose a cosine series up to third order $$ U_\phi(\phi) = \sum_{n=1}^3 k_{\phi, i} (1 + \cos(n\phi - \phi_{0, i})). $$ where we took the values for the force constants $k_{\phi, i}$ and phase shifts $\phi_{0, i}$ from the Amber03 force field [3]. The non-bonded repulsion has the form $$ U_r(x) = \varepsilon\left(\frac{x}{\sigma}\right)^{12}. $$ We computed the pairwise parameters $\varepsilon_{ij} = \sqrt{\varepsilon_{ii}\varepsilon_{jj}}$ and $\sigma_{ij} = \frac{\sigma_{ii} + \sigma_{jj}}{2}$ via the Lorentz-Berthelot combining rules [4] from the Amber03 parameters.
We now construct this prior potential in chemtrain. First, we select the corresponding potential terms, i.e., harmonic bond and angle terms, cosine dihedral angle terms, and repulsive nonbonded terms in the correct space.
prior_energy = prior.init_prior_potential(displacement_fn, nonbonded_type="repulsion")
Following, we load the potential parameters.
with open(base_path / "alanine_heavy.toml") as f:
print(f.read())
force_field = prior.ForceField.load_ff(base_path / "alanine_heavy.toml")
[bonded]
bondtypes = """
# i, j, b0, kb
C, CH3,0.15172,271037.3
C, O,0.12325,476729.7
C, N,0.13359,416066.4
C, CA,0.15445,277737.0
CA, CH3,0.15370,268630.6
CA, N,0.14683,290439.9
CH3, N,0.14592,287462.0
"""
angletypes = """
# i, j, k, th0, kth
CH3, C, O,119.896, 0.319
CH3, C, N,116.911, 0.305
N, C, O,122.485, 0.338
C, N, CA,124.654, 0.247
CH3, CA, N,108.107, 0.260
C, CA, N,113.044, 0.231
C, CA, CH3,111.665, 0.230
CA, C, O,120.259, 0.320
CA, C, N,117.381, 0.301
C, N, CH3,124.797, 0.245
"""
dihedraltypes = """
# i, j, k, l, phase, kd pn
C, N, CA, C, 0.00, 4.251, 1
C, N, CA, C, 180.00, 1.444, 2
C, N, CA, C, 0.00, 0.945, 3
N, CA, C, N, 180.00, 2.861, 1
N, CA, C, N, 180.00, 6.082, 2
N, CA, C, N, 180.00, 1.931, 3
CA, N, C, CH3, 180.00, 10.460, 2
CA, N, C, O, 180.00, 10.460, 2
C, N, CA, CH3, 180.00, 1.480, 1
C, N, CA, CH3, 180.00, 3.697, 2
C, N, CA, CH3, 180.00, 0.950, 3
CH3, CA, C, N, 180.00, 3.257, 1
CH3, CA, C, N, 180.00, 0.275, 2
CH3, CA, C, N, 180.00, 0.234, 3
CA, C, N, CH3, 180.00, 10.460, 2
CH3, N, C, O, 180.00, 10.460, 2
"""
[nonbonded]
# No non-bonded interaction
atomtypes = """
# name, species, mass, sigma, epsilon
CH3, 0, 15.035,3.39967e-01,3.59824e-01
C, 1, 12.011,3.39967e-01,3.59824e-01
O, 2, 15.999,2.95992e-01,8.78640e-01
N, 3, 14.007,3.25000e-01,7.11280e-01
CA, 4, 13.019,3.39967e-01,3.59824e-01
"""
Finally, we must define the index sets for the bonds $\mathcal B$, angles $\mathcal A$, and dihedral angles $\mathcal D$. Luckily, we do not have to gather the correct indices by hand. Instead, chemtrain allows automatically identifying these indices by traversing a molecular graph, e.g., constructed from the mdtraj package [5].
top = mdtraj.load_topology("../_data/alanine_heavy_2_7nm.gro")
_mapping = force_field.mapping(by_name=True)
def mapping(name="", residue="", **kwargs):
if residue == "NME" and name =="C":
return _mapping(name="CH3", **kwargs)
if name == "CB":
return _mapping(name="CH3", **kwargs)
else:
return _mapping(name=name, **kwargs)
topology = prior.Topology.from_mdtraj(top, mapping)
species = topology.get_atom_species()
masses, *_ = force_field.get_nonbonded_params(species)[0].T
Model definition#
With the prior potential defined, we can now construct the learnable difference $\Delta U_\theta$. We select the DimeNet++ graph neural network architecture [6], shipped with chemtrain.
# We need an example graph (neighbor list) to determine the number
# of nodes, edges, angles, etc.
mlp_init = {
'b_init': hk.initializers.Constant(0.),
'w_init': layers.OrthogonalVarianceScalingInit(scale=1.)
}
init_fn, gnn_energy_fn = neural_networks.dimenetpp_neighborlist(
displacement_fn, r_cut, n_species, r_init, nbrs_init,
embed_size=32, init_kwargs=mlp_init,
)
# Create the parametrizable energy function and an initial
# parametrization
key, split = random.split(key)
init_params = init_fn(
split, r_init, neighbor=nbrs_init, species=species
)
def energy_fn_template(energy_params):
prior_energy_fn = prior_energy(topology, force_field)
def energy_fn(pos, neighbor, **dynamic_kwargs):
gnn_energy = gnn_energy_fn(
energy_params, pos, neighbor, species=species,
**dynamic_kwargs
)
prior_energy = prior_energy_fn(pos, neighbor=neighbor)
return gnn_energy + prior_energy
return energy_fn
Reference Data#
We use the reference data from the original paper [1]. This reference data consists of $5\times10^{5}$ conformations with corresponding forces, subsampled every $200~\text{fs}$ from a $100~\text{ns}$ all-atomistic simulation of alanine-dipeptide in TIP3P water.
# Download if not present
position_url = "https://drive.usercontent.google.com/download?id=1yKVHiI8y7ZNzyduh8bosR6YKScLyFezU&export=download&confirm=t&uuid=cff71a05-a45a-446e-bf2f-17f62e84263f"
force_url = "https://drive.usercontent.google.com/download?id=1JhRQcZ3tE2w-mLqTGN0JHJQst5uZijJx&export=download&confirm=t&uuid=ad4f279f-18b7-4b65-a144-ab1115110549"
forces_path = "../_data/forces_heavy_100ns.npy"
positions_path = "../_data/positions_heavy_100ns.npy"
if not Path(forces_path).exists():
request.urlretrieve(force_url, forces_path)
if not Path(positions_path).exists():
request.urlretrieve(position_url, positions_path)
force_dataset = preprocessing.get_dataset(forces_path)
position_dataset = preprocessing.get_dataset(positions_path)
if fractional:
position_dataset = preprocessing.scale_dataset_fractional(
position_dataset, box
)
Simulation#
As outlined in the Relative Entropy example, estimating the gradients of the relative entropy requires samples from the current coarse-grained ensemble. Moreover, we want to compare the learned models in predicting the distributions of backbone dihedral angles. Thus, we need to simulate CG alanine dipeptide based on the neural network and prior potential. However, we do not set up a single long simulation. Instead, we set up $100$ parallel and shorter simulations to accelerate the sampling. These simulations start from conformations randomly selected from the reference data without replacement.
dt = 0.002
n_chains = 100
gamma = 100.
key, split = random.split(key)
selection = random.choice(
split, jnp.arange(position_dataset.shape[0]), shape=(n_chains,), replace=False)
r_init = position_dataset[selection, ...]
init_ref_state, sim_template = sampling.initialize_simulator_template(
simulate.nvt_langevin, shift_fn=shift_fn, nbrs=nbrs_init,
init_with_PRNGKey=True, extra_simulator_kwargs={"kT": kT, "gamma": gamma, "dt": dt}
)
key, split = random.split(key)
reference_state = init_ref_state(
split, r_init,
energy_or_force_fn=energy_fn_template(init_params),
init_sim_kwargs={"mass": masses, "neighbor": nbrs_init}
)
Relative Entropy Minimization#
With reference data, potential model, and simulation routine set up, we can now instantiate the REM algorithm. We only use a subset of $80~%$ of the data for training. We also set the reweighting ratio to issue a new trajectory after every update. Nevertheless, we tune the step size such that at least $25~%$ effective samples remain after each update.
re_initial_lr = 0.003
re_epochs = 300
re_used_dataset_size = 400000
t_sample = 0.5
total_time = 110.
t_eq = 10.
re_timings = sampling.process_printouts(
time_step=dt, total_time=total_time,
t_equilib=t_eq, print_every=t_sample
)
lr_schedule = optax.exponential_decay(re_initial_lr, re_epochs, 0.01)
optimizer = optax.chain(
optax.scale_by_adam(0.1, 0.4),
optax.scale_by_schedule(lr_schedule),
optax.scale_by_learning_rate(1.0)
)
relative_entropy = trainers.RelativeEntropy(
init_params, optimizer, reweight_ratio=1.1,
energy_fn_template=energy_fn_template)
relative_entropy.add_statepoint(
position_dataset[:re_used_dataset_size, ...],
energy_fn_template, sim_template, neighbor_fn,
re_timings, state_kwargs, reference_state,
reference_batch_size=re_used_dataset_size,
vmap_batch=n_chains, resample_simstates=True)
relative_entropy.init_step_size_adaption(0.25)
if os.environ.get("RM_TRAINING", "False").lower() == "true":
# Save the training log
with open("../_data/output/alanine_dipeptide_rm_training.log", "w") as f:
with contextlib.redirect_stdout(f), contextlib.redirect_stderr(f):
start = time.time()
relative_entropy.train(300)
print(f"Total training time: {(time.time() - start) / 3600 : .1f} hours")
relative_entropy.save_energy_params("../_data/output/alanine_dipeptide_re_params.pkl", '.pkl')
relative_entropy.save_trainer("../_data/output/alanine_dipeptide_re_trainer.pkl", '.pkl')
relative_entropy = onp.load("../_data/output/alanine_dipeptide_re_trainer.pkl", allow_pickle=True)
relative_entropy_params = tree_util.tree_map(
jnp.asarray, onp.load("../_data/output/alanine_dipeptide_re_params.pkl", allow_pickle=True)
)
with open("../_data/output/alanine_dipeptide_rm_training.log") as f:
print(f.read())
Plotting the change of relative entropy and the gradient norm indicates convergence of the algorithm for $300$ epochs.
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(9, 4), layout="constrained")
ax1.plot(relative_entropy["delta_re"][0])
ax1.set_xticks(ticks=range(0, re_epochs + 1, 50))
ax1.set_xlabel("Epoch")
ax1.set_ylabel("RE Loss")
ax2.plot(relative_entropy["gradient_norm_history"])
ax2.set_xticks(ticks=range(0, re_epochs + 1, 50))
ax2.set_xlabel("Epoch")
ax2.set_ylabel("Gradient Norm")
Text(0, 0.5, 'Gradient Norm')

Force Matching#
As a reference, we train an instance of the potential model via FM. Similar to REM, we only consider a subset of the data for training. However, mainly to prevent overfitting, we need additional data for validation. Hence, the part of data we can use for training is smaller.
fm_epochs = 100
fm_used_dataset_size = 500000
fm_train_ratio = 0.7
fm_val_ratio = 0.1
fm_batch_per_device = 500 // len(jax.devices())
fm_batch_cache = 50
fm_initial_lr = 0.0003
lrd = int(fm_used_dataset_size / fm_batch_per_device / len(jax.devices()) * fm_epochs)
lr_schedule = optax.exponential_decay(fm_initial_lr, lrd, 0.01)
fm_optimizer = optax.chain(
optax.scale_by_adam(),
optax.scale_by_schedule(lr_schedule),
optax.scale_by_learning_rate(1.0)
)
force_matching = trainers.ForceMatching(
init_params, fm_optimizer, energy_fn_template, nbrs_init,
batch_per_device=fm_batch_per_device,
batch_cache=fm_batch_cache,
)
force_matching.set_datasets({
'R': position_dataset[:fm_used_dataset_size, ...],
'F': force_dataset[:fm_used_dataset_size, ...]
}, train_ratio=fm_train_ratio
)
if os.environ.get("FM_TRAINING", "False").lower() == "true":
# Save the training log
with open("../_data/output/alanine_dipeptide_fm_training.log", "w") as f:
with contextlib.redirect_stdout(f), contextlib.redirect_stderr(f):
print(f"Visible devices: {jax.devices()}")
start = time.time()
force_matching.train(fm_epochs)
print(f"Total training time: {(time.time() - start) / 3600 : .1f} hours")
force_matching.save_energy_params("../_data/output/alanine_dipeptide_fm_params.pkl", '.pkl', best=False)
force_matching.save_trainer("../_data/output/alanine_dipeptide_fm_trainer.pkl", '.pkl')
force_matching = onp.load("../_data/output/alanine_dipeptide_fm_trainer.pkl", allow_pickle=True)
force_matching_params = tree_util.tree_map(
jnp.asarray, onp.load("../_data/output/alanine_dipeptide_fm_params.pkl", allow_pickle=True)
)
with open("../_data/output/alanine_dipeptide_fm_training.log") as f:
print(f.read())
Plotting the training and validation loss indicates convergence after approximately $50$ epochs. Further training improves the validation loss only slightly, but does not lead to overfitting. Therefore, we save the final model with $100$ training epochs.
fig, ax = plt.subplots(1, 1, figsize=(5, 4), layout="constrained")
ax.plot(force_matching["train_losses"], label="Training")
ax.plot(force_matching["val_losses"], label="Validation")
ax.set_xticks(ticks=range(0, fm_epochs + 1, 25))
ax.set_xlabel("Epoch")
ax.set_ylabel("MSE Force Loss")
ax.legend()
<matplotlib.legend.Legend at 0x7f1d102b8490>

Force Matching and Relative Entropy Minimization#
Due to the expense of training with relative entropy minimization, we train a third model from an improved initial state. As improved initial state, we use the FM trained model with the lowest validation loss.
rm_post_epochs = 50
rm_post_initial_lr = 0.0005
lr_schedule = optax.exponential_decay(rm_post_initial_lr, rm_post_epochs, 0.01)
optimizer = optax.chain(
optax.scale_by_adam(0.1, 0.4),
optax.scale_by_schedule(lr_schedule),
optax.scale_by_learning_rate(1.0)
)
rm_post_fm = trainers.RelativeEntropy(
force_matching_params, optimizer, reweight_ratio=1.1,
energy_fn_template=energy_fn_template)
rm_post_fm.add_statepoint(
position_dataset[:re_used_dataset_size, ...],
energy_fn_template, sim_template, neighbor_fn,
re_timings, state_kwargs, reference_state,
reference_batch_size=re_used_dataset_size,
vmap_batch=n_chains, resample_simstates=True)
rm_post_fm.init_step_size_adaption(0.25)
/home/paul/chemtrain_rerun_ad/chemtrain/ensemble/reweighting.py:777: UserWarning: Propagation function is not safe by default. Do not forget to use the wrapper around the compute function to ensure that the neighborlist does not overflow.
warnings.warn(
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
Time for trajectory initialization 0: 2.6623266140619912 mins
[Step size] Use 7 iterations for 10 interior points.
if os.environ.get("FM_RM_TRAINING", "False").lower() == "true":
# Save the training log
with open("../_data/output/alanine_dipeptide_fm+rm_training.log", "w") as f:
with contextlib.redirect_stdout(f), contextlib.redirect_stderr(f):
start = time.time()
rm_post_fm.train(rm_post_epochs)
print(f"Total training time: {(time.time() - start) / 3600 : .1f} hours")
rm_post_fm.save_energy_params("../_data/output/alanine_dipeptide_fm+rm_params.pkl", '.pkl')
rm_post_fm.save_trainer("../_data/output/alanine_dipeptide_fm+rm_trainer.pkl", '.pkl')
rm_post_fm = onp.load("../_data/output/alanine_dipeptide_fm+rm_trainer.pkl", allow_pickle=True)
rm_post_fm_params = tree_util.tree_map(
jnp.asarray, onp.load("../_data/output/alanine_dipeptide_fm+rm_params.pkl", allow_pickle=True)
)
with open("../_data/output/alanine_dipeptide_fm+rm_training.log") as f:
print(f.read())
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(9, 4), layout="constrained")
ax1.plot(rm_post_fm["delta_re"][0])
ax1.set_xticks(ticks=range(0, rm_post_epochs + 1, 10))
ax1.set_xlabel("Epoch")
ax1.set_ylabel("RE Loss")
ax2.plot(rm_post_fm["gradient_norm_history"])
ax2.set_xticks(ticks=range(0, rm_post_epochs + 1, 10))
ax2.set_xlabel("Epoch")
ax2.set_ylabel("Gradient Norm")
Text(0, 0.5, 'Gradient Norm')

Evaluation#
With both trained models, we perform a series of simulations to evaluate the alignment of both models with the reference data. To reduce the total running time, we again run $100$ shorter simulations, which still correspond to a total time of $100~\text{ns}$.
eval_total_time = 2100.
eval_t_eq = 100.
eval_t_sample = .5
eval_timings = sampling.process_printouts(
time_step=dt, total_time=eval_total_time,
t_equilib=eval_t_eq, print_every=eval_t_sample
)
trajectory_generator = sampling.trajectory_generator_init(
sim_template, energy_fn_template, eval_timings)
trajectory_generator = jax.vmap(
functools.partial(trajectory_generator, **state_kwargs), (0, None)
)
if os.environ.get("EVALUATION", "False").lower() == "true":
all_parameters = util.tree_stack((
relative_entropy_params,
force_matching_params,
rm_post_fm_params
))
t_start = time.time()
all_traj_states = trajectory_generator(all_parameters, reference_state)
assert not jnp.any(all_traj_states.overflow), (
'Neighborlist overflow during trajectory generation. '
'Increase capacity and re-run.'
)
print(f'Total runtime: {(time.time() - t_start) / 3600 :.1f} hours')
(re_traj_state, fm_traj_state, fm_rm_traj_state) = util.tree_unstack(all_traj_states)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
/home/paul/miniconda3/envs/chemtrain_rerun_AD/lib/python3.11/site-packages/jax/_src/numpy/lax_numpy.py:166: UserWarning: Explicitly requested dtype float64 requested in asarray is not available, and will be truncated to dtype float32. To enable more dtypes, set the jax_enable_x64 configuration option or the JAX_ENABLE_X64 shell environment variable. See https://github.com/google/jax#current-gotchas for more.
return asarray(x, dtype=self.dtype)
Total runtime: 4.4 hours
def postprocess_fn(positions):
# Compute the dihedral angles
dihedral_idxs = jnp.array([[1, 3, 4, 6], [3, 4, 6, 8]]) # 0: phi 1: psi
batched_dihedrals = jax.vmap(
custom_quantity.dihedral_displacement, (0, None, None)
)
dihedral_angles = batched_dihedrals(positions, displacement_fn, dihedral_idxs)
return dihedral_angles.T
if os.environ.get("EVALUATION", "False").lower() == "true":
ref_phi, ref_psi = postprocess_fn(position_dataset)
fm_phi, fm_psi = postprocess_fn(fm_traj_state.trajectory.position)
re_phi, re_psi = postprocess_fn(re_traj_state.trajectory.position)
fm_rm_phi, fm_rm_psi = postprocess_fn(fm_rm_traj_state.trajectory.position)
onp.savez(
"../_data/output/alanine_dipeptide_dihedral_angles.npz",
ref_phi=ref_phi, ref_psi=ref_psi,
fm_phi=fm_phi, fm_psi=fm_psi,
re_phi=re_phi, re_psi=re_psi,
fm_rm_phi=fm_rm_phi, fm_rm_psi=fm_rm_psi
)
results = onp.load("../_data/output/alanine_dipeptide_dihedral_angles.npz")
ref_phi=results["ref_phi"]
ref_psi=results["ref_psi"]
fm_phi=results["fm_phi"]
fm_psi=results["fm_psi"]
re_phi=results["re_phi"]
re_psi=results["re_psi"]
fm_rm_phi=results["fm_rm_phi"]
fm_rm_psi=results["fm_rm_psi"]
def plot_1d_dihedral(ax, angles, labels, bins=60, degrees=True,
xlabel='$\phi$ in deg', ylabel=True):
"""Plot 1D histogram splines for a dihedral angle. """
color = ['#368274', '#0C7CBA', '#C92D39', 'k']
line = ['-', '-', '-', '--']
n_models = len(angles)
for i in range(n_models):
if degrees:
angles_conv = angles[i]
hist_range = [-180, 180]
else:
angles_conv = onp.rad2deg(angles[i])
hist_range = [-onp.pi, onp.pi]
# Compute the histogram
hist, x_bins = jnp.histogram(angles_conv, bins=bins, density=True, range=hist_range)
width = x_bins[1] - x_bins[0]
bin_center = x_bins + width / 2
ax.plot(
bin_center[:-1], hist, label=labels[i], color=color[i],
linestyle=line[i], linewidth=2.0
)
ax.set_xlabel(xlabel)
if ylabel:
ax.set_ylabel('Density')
return ax
def plot_histogram_free_energy(ax, phi, psi, kbt, degrees=True, ylabel=False, title=""):
"""Plot 2D free energy histogram for alanine from the dihedral angles."""
cmap = plt.get_cmap('viridis')
if degrees:
phi = jnp.deg2rad(phi)
psi = jnp.deg2rad(psi)
h, x_edges, y_edges = jnp.histogram2d(phi, psi, bins=60, density=True)
h = jnp.log(h) * -(kbt / 4.184)
x, y = onp.meshgrid(x_edges, y_edges)
cax = ax.pcolormesh(x, y, h.T, cmap=cmap, vmax=5.25)
ax.set_xlabel('$\phi$ [rad]')
if ylabel:
ax.set_ylabel('$\psi$ [rad]')
ax.set_title(title)
return ax, cax
Plotting the dihedral angle distributions reveals that both FM and REM-trained models can identify the preferred torsional states of alanine-dipeptide. However, the REM model reproduces the relative preference between the states much better.
labels = ["FM", "RM", "FM + RM", "AA Reference"]
fig, (ax1, ax2) = plt.subplots(1, 2, layout="constrained", figsize=(9, 3), sharey=True)
ax1 = plot_1d_dihedral(ax1, [fm_phi, re_phi, fm_rm_phi, ref_phi], labels, xlabel="$\phi\ [deg]$")
ax2 = plot_1d_dihedral(ax2, [fm_psi, re_psi, fm_rm_psi, ref_psi], labels, xlabel="$\psi\ [deg]$", ylabel=False)
fig.legend(labels, ncols=len(labels), bbox_to_anchor=(0.5, 1.01), loc="lower center")
fig.savefig("../_data/output/alanine_dipeptide_1D_dihedral_angles.pdf", bbox_inches='tight')

Plotting the free energy surface of the backbone dihedral angles indicates similar results. Both models can identify the regions of low free energy. However, only the REM model correctly predicts the depth of these regions.
labels = ["AA Reference"]
fig, (ax1, ax2, ax3, ax4) = plt.subplots(1, 4, layout="constrained", figsize=(9, 3), sharey=True)
ax1, _ = plot_histogram_free_energy(ax1, ref_phi, ref_psi, kT, ylabel=True, title="AA Reference")
ax2, _ = plot_histogram_free_energy(ax2, fm_phi, fm_psi, kT, title="Force Matching")
ax3, _ = plot_histogram_free_energy(ax3, re_phi, re_psi, kT, title="Relative Entropy")
ax4, cax = plot_histogram_free_energy(ax4, fm_rm_phi, fm_rm_psi, kT, title="FM and RM")
cbar = fig.colorbar(cax)
cbar.set_label('Free Energy (kcal/mol)')
fig.savefig("../_data/output/alanine_dipeptide_free_energy_dihedral_angles.pdf", bbox_inches='tight')
